

Vanitas Still Life by Harmen Steenwyck, Kunsthistorisches Museum, Vienna, Austria (Oct 2024)
I love surprises in paintings…the little things that you only notice if you look at a painting up close so when I started to notice the butterflies, lizards, ladybugs, and dragonflies in Dutch/Flemish paintings from the 17th century, I was absolutely thrilled. It’s not just about the pretty flowers; it’s about the little surprises I find only when I stop and take my time in front of an art piece.
This is the second blog post in my Nope, Nope, Nope…Oh, That One series on making art more accessible to my friend Sandy, and anyone else who gets completely overwhelmed when walking into an art museum.
When I first saw the piece above, Vanitas Still Life by Harmen Steenwyck, my first thought was, “there’s a lot going on…this is absolute chaos!”
But then I zoomed in and focused on the butterfly, appreciating its delicacy. I looked around for other butterflies or creepy crawly critters, and seeing none, went to the next thing that caught my eye…the corncob with its bright yellow kernels and chewed away cob.
I continued to look around the painting glancing past the writing on the book but appreciating the how real the bent pages looked before noticing the blue and white China inkwell that could be either Dutch or Asian.
I loved the creepiness of the skull. This is another thing I now look for in Dutch/Flemish paintings because there was a time when they included skulls to signify mortality. I’m just wondering how an artist just has random things like skulls lying around their studio or salon.
In most florals, the first thing I notice are the flowers, but in this particular piece, the gorgeous and realistic flower arrangement isn’t the central focus. I still think it’s amazing and I aspire to paint flowers like these (I’m a long way from it at this point).
Let’s play a grown-up game of Hidden Pictures, like we did in the Highlights magazine when we were kids, and look for all of the hidden objects. Kind of an I spy with my little eyes game. I’ll share what I see and what caught my eye. Let me know if you see something different.
And next time you are at a museum, and you feel overwhelmed, take a look at the old floral arrangements and see if the artist included little surprises.

Flowers in a Glass Vase by Ambrosius Bosschaert, the elder, Canter Center, Stanford, California (Feb 2026)
Besides the gorgeous black and white (possibly parrot) tulip on the left, I spied with my little eyes a butterfly and a dragonfly.

Flowers in a Glass Vase by Ambrosius Bosschaert, the elder, Canter Center, Stanford, California (Feb 2026)
Just in case you couldn’t see it, I zoomed in on the dragonfly.

Flowers in a Glass Vase by Ambrosius Bosschaert, the elder, Canter Center, Stanford, California (Feb 2026)
And the butterfly. But check out the stem of the white tulip while you are here, and then the lusciousness of the pink flower. I can practically feel it.

Flowers and Fruits on a Marble Bench, by Jan van Huysym, Musee des Beaux-Arts, Lyon, France (Oct 2025)
This one is just crazy. What the hell is going on with the pineapple? How is this arrangement even held together?

Flowers and Fruits on a Marble Bench, by Jan van Huysym, Musee des Beaux-Arts, Lyon, France (Oct 2025)
Zoom in on the butterfly…I think it’s very cool. But the walnut? Why does it look like it’s dripping? And why the random cherries, with one looking slightly bruised? Would these be modern day sexual emojis?
I do love how realistic the skin of the green squash is. I can imagine feeling the texture of it as I run an imaginary hand over it. I just saw the corn to the left of the peaches and above the rotting (?) squash. And something I hadn’t noticed before…a bee!

Couronne et couple des fleurs by Jan II Brueghel, Musee des Beaux-Arts, Strasbourg, France (Sept 2025)
What drew me to this one was the vase…it screamed Beauty and the Beast, so of course I had to take a closer look.

Couronne et couple des fleurs by Jan II Brueghel, Musee des Beaux-Arts, Strasbourg, France (Sept 2025)
Once I moved closer to look at this incredible vase that really does look like Lumière, I saw the beetle. I almost missed the little gal. She looked like some petals or leaves that had dropped from the arrangement. The flowers are pretty but they are the least interesting part of this painting to me.
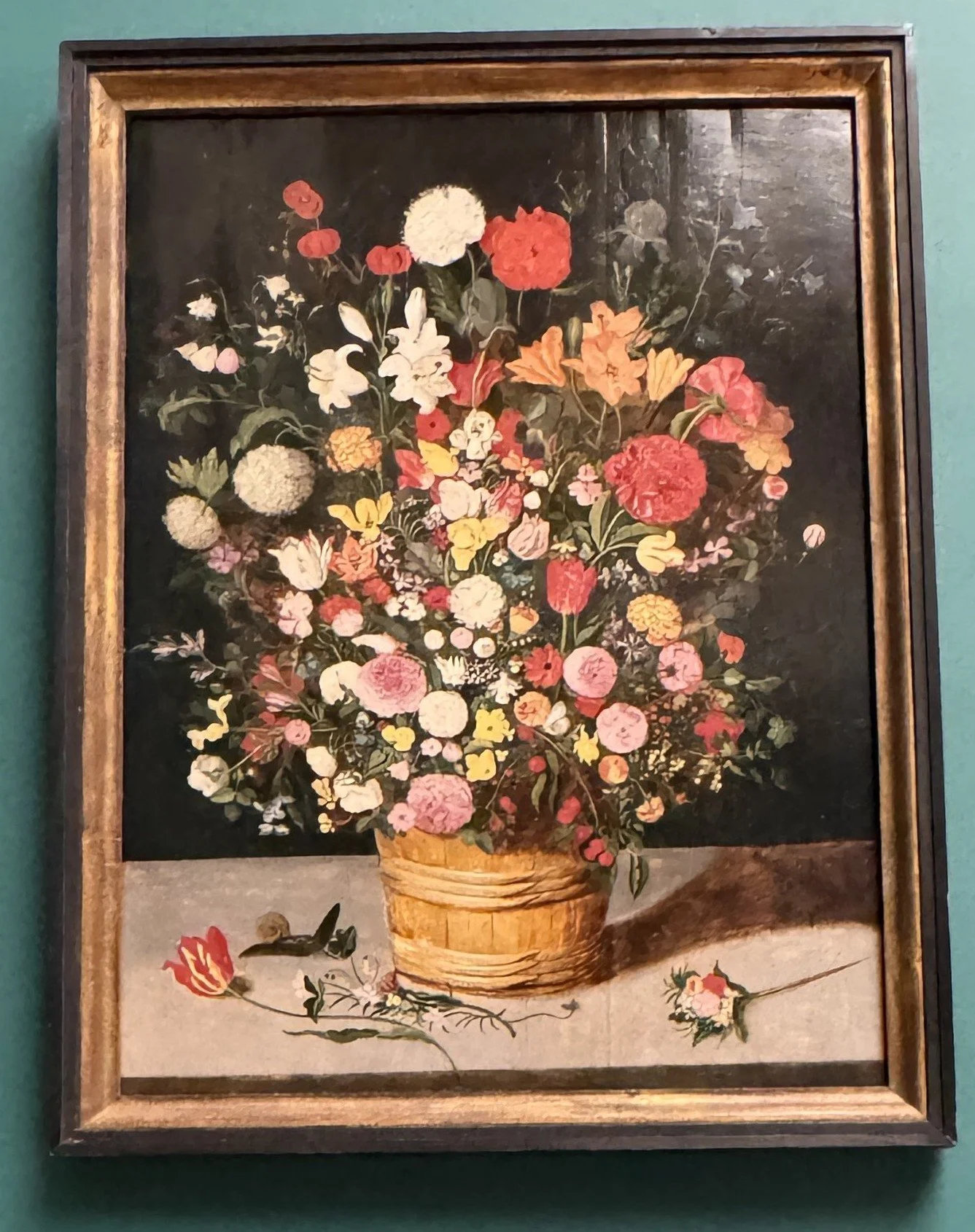
Bouquet des fleurs by a follower of Jan I Brueghel, Musee des Beaux-Arts, Strasbourg, France (Sept 2025)
This one is hung directly above the previous piece at the Musee des Beaux-Arts in Strasbourg, a lovely and petite fine arts museum in the Palais de Rohan. This is another chaotic arrangement but there are some fun surprises.

Bouquet des fleurs by a follower of Jan I Brueghel, Musee des Beaux-Arts, Strasbourg, France (Sept 2025)
Check out the snail! There’s no way the slug part is going to fit into that shell. While the snail isn’t very realistic, the parrot tulip is gorgeous and I do appreciate the wooden bucket used to hold all of the flowers which brings me to another one of my favorite things to look at in these pieces.

Fleurs dans un grand vase, d’orfevrerie by Abraham Bruegher, Musee des Beaux-Arts, Dijon, France (Oct 2025)
The vases! The title of this one in English is Flowers in a Large, Finely Wrought Vase.

Bouquet des fleurs by Jan van Huysum, Musee des Beaux-Arts, Strasbourg, France (Sept 2025)
The grapes may look like glass balls but there is no doubt that this is a terracotta pot with two cheeky babes. I know I’m trying to keep you focused on the vase, but check out how realistic the white and pink carnation is. And look! A white butterfly.

Vase de fleurs sur fond de parc avec statue by Jan van Huysum, Musee des Beaux-Arts, Dijon, France (Oct 2025)
I love how unhappy this cherub is on the vase. It’s pretty creepy but then there are two white butterflies and a caterpillar, and of course the beautiful flowers, to offset this creepiness. Jan really knows how to pull together a compelling composition full of delights.
Now for amateur hour!

I recently started painting with gouaches and acrylics and did a two-page floral arrangement. I’m pretty proud of it given it was the first time I tried something like this. But I’ve just now realized that it’s missing something (or a couple of somethings). I think it’s time to go back and add some hidden surprises.
But seriously, the next time you go to a museum, look for the floral arrangements. Look at them up close to see if the artist has added some surprises or painted the flowers in an interesting vase. And then look at the flowers and see if you can identify any of them. I’m confident you will find something to delight you.
Extra credit: look for the women artists.
In the 17th and 18th centuries, it was considered “unseemly” for women to paint anything other than florals or botanicals. Artists like Rachel Ruysch, Clara Peeters, and Maria Sibylla Merian did some amazing work and should not be overlooked.

Rijksmuseum, Amsterdam, Netherlands (May 2023)
After my son Adam graduated college in May 2023, before our Etruscan adventures I mentioned in my first post, we went to Amsterdam. While in Amsterdam, since we both LOVE museums and art, we went to the Rijksmuseum, a truly amazing museum that we both long to return to as it deserves a week, not an afternoon.
The building itself is gorgeous including stunning stained-glass windows, exquisite mosaic tile floors — including portrait medallions set right into the floor that most visitors walk right over — and a lavish garden, both formal and botanical. It was purpose-built as a museum to house its collection of 1 million objects dedicated to arts, crafts, and history from 1200 to 2000.

Rijksmuseum, Amsterdam, Netherlands (May 2023)
As we walked through in a bit of a daze from jet lag and sheer overwhelm, we were both surprised to see a placard next to a series of botanicals and florals by women artists, intentionally displayed throughout the museum. The museum was recognizing, and attempting to remedy, the fact that women artists had contributed greatly to Dutch cultural history and were not effectively represented. The Rijksmuseum had also begun a research program to identify women’s contributions to its collection and at that point had identified 29,311 objects by 2,908 women, including 158 of the museum’s 7,173 paintings.
We need to see more of this. I’m reading The Story of Art Without Men by Katy Hessel to learn more about women in art and how we as a society are attempting to do a better job of adding back to history those who have been intentionally overlooked. I’ll talk more about this in a future post.

About the Author
Terri Hanson Mead is the multi-award winning author of Piloting Your Life, Managing Partner of Solutions2Projects, LLC, travel journalist and vlogger with her husband Zeke (Zeke and Terri Adventures), Stanford Continuing Studies Instructor (Navigating Midlife for Women), and an advocate for women through all of her platforms including YouTube, Instagram, TikTok, and this blog. Terri, the mother of a college senior and recent college graduate (pursuing his master’s in philosophy at SF State), is based in Redwood City, CA and in her spare time, loves to travel, cook, play tennis, and fly helicopters around the San Francisco Bay Area, especially under the Golden Gate Bridge. Oh, and she will never pass up a glass of good bubbly or fun cocktail (even if she wants a good night’s sleep!)!